
Viewpoint: FDA regulation of mobile medical apps
Mobile apps are increasingly used in health care to promote wellness, treat and diagnose disease, aid clinical decision-making, and manage patient care in hospitals and homes.
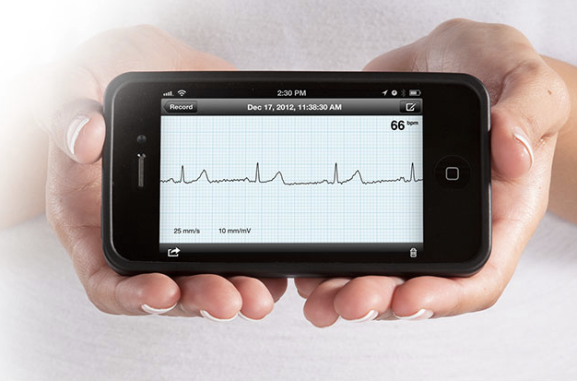
Alivecor is one of thousands of new mobile health apps. It lets you measure your heart rate with an iPhone. Alivecor photo
Jeffrey Shuren MD JD; Bakul Patel MS MBA; Scott Gottlieb MD, JAMA Network July 2, 2018
Historically, health care has been slow to implement disruptive technology tools that have transformed other areas of commerce and daily life. One factor that has been cited is uncertainty surrounding regulation that accompanies medical products, and how US Food and Drug Administration (FDA) regulations may apply to software platforms. There also are questions in the marketplace about the clinical validity and utility of certain mobile tools.
Efficient regulation can help promote adoption of mobile medical apps. FDA determination that a product developer or manufacturer has met the high regulatory standard for demonstrating clinical benefit and safety (when agency clearance or approval of the app is required) can increase consumer confidence in that technology.1 that makes it clear that not all these tools are subject to FDA regulation. The agency oversees most mobile apps that are intended to treat, diagnose, cure, mitigate, or prevent disease or other conditions as medical devices under federal statute. Not all mobile apps meet these characteristics. But for devices evaluated, the policies must continue to empower patients and clinicians and facilitate innovation, including by creating regulatory frameworks that give patients and clinicians confidence in the app’s performance and reliability.
Mobile medical apps may help overcome the siloed, episodic, reactive nature of US health care, whereby patients seek care only after potentially costly health complications occur, and physicians are only reimbursed for expensive in-person office visits that may not reflect the day-to-day reality of the patient experience of living with complex chronic conditions.
Increased demand for mobile medical apps could encourage greater integration of apps with electronic health records (EHRs), potentially allowing clinicians and patients to better manage complex health conditions based on near real-time feedback loops documenting patients’ feeling or function. Structured data flows from EHRs and wearable devices could also be used to better inform regulatory decision-making related to drug and device safety or efficacy.
The FDA position is that efficient regulation of mobile medical apps should be tailored to their potential benefits and risks.
After careful consideration, the agency has released guidance2 that makes it clear that not all these tools are subject to FDA regulation. The agency oversees most mobile apps that are intended to treat, diagnose, cure, mitigate, or prevent disease or other conditions as medical devices under federal statute. Not all mobile apps meet these characteristics. But for devices evaluated, the policies must continue to empower patients and clinicians and facilitate innovation, including by creating regulatory frameworks that give patients and clinicians confidence in the app’s performance and reliability.
However, the regulatory framework enacted by Congress in 1976, and incrementally improved since then, is not well suited for software-based technologies, including mobile apps, what FDA and other regulators call “software as a medical device” (SaMD). Congress’ regulatory framework was designed for hardware-based technologies. For these devices, developers may only modify products every few months to years, and much can be learned about the technology from its design, composition, and bench testing. The effects of these products on patients tend to be readily observable, and knowledge generated about one product often can be applied to others in the same category to expedite regulatory decision-making. For example, the effect of a synthetic valve replacement device for aortic stenosis on cardiac output can be readily measured and information that becomes known about the performance and failure modes of one such device can be readily applied to the evaluation of a similar synthetic valve.
This regulatory framework uses a risk-based approach to ensure that all devices on the US market provide a reasonable assurance of safety and effectiveness. What a developer must do for its product to meet this standard depends on the risks posed to patients should the device fail to perform as intended. Makers of low-risk devices, such as bandages and eyeglasses, must provide truthful, nonmisleading labeling, implement a system to ensure product and manufacturing quality, report to FDA serious adverse events, deaths, and malfunctions associated with their product, and take appropriate action if and when problems arise.
Makers of moderate- and high-risk devices, such as magnetic resonance imaging scanners and cardiac pacemakers, generally must also gather nonclinical, and sometimes clinical, evidence to show they meet the standard and include it in a premarket submission to FDA for review to determine whether to authorize marketing of the technology. Modifications that could affect the safety or effectiveness of the device undergo a similar premarket review.
By contrast, developers of SaMD, such as clinical decision support software designed to analyze computed tomography results that notifies clinicians of a potential stroke in their patients, can modify their products in response to performance in clinical settings and user feedback every few weeks to months, and little to nothing can be learned about the technology by just reviewing the software code. The influence of apps on patients may be indirect, and knowledge about one software program generally cannot be applied to other programs with the same intended use.
In contrast, SaMD products offer unique opportunities such as addressing malfunctions quickly and efficiently through software updates to minimize adverse events, and directly capturing the effects involving patients outside of the clinical setting, enabling enhanced near-real-time patient engagement and learning. SaMD may also present new challenges, such as addressing cybersecurity vulnerabilities.
The traditional application of FDA’s longstanding regulatory framework can stifle the development of, and access to, new and improved SaMD while providing limited patient safeguards. To meet its core mission of promoting and protecting public health, it is important for FDA to create a regulatory framework for SaMD that recognizes the distinctive aspects of digital health technology, including its clinical promise, unique user interface, and compressed commercial cycles for new product introductions and modifications.
To address these challenges as well as the needs of FDA’s customers for greater clarity about the agency’s regulatory approach, in 2011 FDA began issuing a series of policy guidance3 to provide market clarity as well as to deregulate many lower-risk functionalities for which active FDA oversight would provide little to no public health value while unnecessarily delaying patient access to potentially beneficial technologies.
These guidance policies addressed those medical device apps for which FDA would continue to actively oversee, called “mobile medical apps,” and those that made general wellness claims, and medical device data systems—technologies that receive, transmit, store, and provide simple displays of information—for which the FDA would not4 and incidents across the total product life cycle for medical devices, including mobile medical apps.
In 2016, in the 21st Century Cures Act Congress amended the Federal Food, Drug, and Cosmetic Act to codify many of these policies and added certain clinical decision support functionalities as no longer being medical devices subject to FDA oversight. The agency also has issued policies to help address cybersecurity vulnerabilities5 and incidents across the total product life cycle for medical devices, including mobile medical apps.
More recently, FDA has led a working group of regulators from several countries under the auspices of the International Medical Device Regulators Forum (IMDRF)6 to establish basic policies for a new, pragmatic, and internationally harmonized regulatory framework for SaMD that better meets patients’ and clinicians’ needs, and the rapid innovation cycles and business models of SaMD developers.
In July 2017, FDA issued a Digital Health Innovation Action Plan7 that described the actions the agency committed to take to fully implement the software provisions of the Cures Act, including to issue new policy on clinical and patient decision support software, establish a dedicated Digital Health Unit in the FDA’s medical device center supported by industry user fee funding, and implement a new regulatory model for digital health technologies consistent with the IMDRF policies.
As part of the latter effort, FDA announced a pilot to create a precertification program under which SaMD developers could be assessed by FDA or an accredited third party for the quality of their software design, testing, and other appropriate capabilities to qualify for a more streamlined premarket review process or in lieu of premarket review depending on the risk of their product while better leveraging postmarket data collection on the device’s safety and effectiveness.
This firm-based approach differs from the agency’s traditional reliance on individual product reviews. Eligible sponsors could engage in more efficient evidence generation by leveraging clinical data from device registries, EHRs, and other electronic health information sources through the National Evaluation System for health Technology (NEST) that is currently under development. The goal of this program is to collaboratively develop a tailored and pragmatic framework that trusts the excellence of organizations, but continually verifies the safety and effectiveness of SaMD.
Through these innovative approaches, FDA seeks to foster technology innovations. At the same time, the agency is committed to providing consumers and clinicians with better information and greater assurances that medical mobile apps and other digital health medical devices that fall within the agency’s regulatory purview are safe and effective.
Source JAMA Network
FDA Regulation of Mobile Medical Apps, Jeffrey Shuren MD JD; Bakul Patel MS, MBA; Scott Gottlieb MD. JAMA. Published online July 2, 2018. doi:10.1001/jama.2018.8832
| References |
|
Also see
FDA wants to encourage development of mobile medical apps with smart regulation in Health Exec
Wellness apps evade the FDA, only to land in court in Wired
FDA removes roadblocks to medical app innovation in Venture Beat